
Abstract
The differentiation of several T- and B-cell effector programs in the immune system is directed by signature transcription factors that induce rapid epigenetic remodelling. Here we report that promyelocytic leukaemia zinc finger (PLZF), the BTB-zinc finger (BTB-ZF) transcription factor directing the innate-like effector program of natural killer T-cell thymocytes1,2, is prominently associated with cullin 3 (CUL3), an E3 ubiquitin ligase previously shown to use BTB domain-containing proteins as adaptors for substrate binding3,4,5,6,7. PLZF transports CUL3 to the nucleus, where the two proteins are associated within a chromatin-modifying complex. Furthermore, PLZF expression results in selective ubiquitination changes of several components of this complex. CUL3 was also found associated with the BTB-ZF transcription factor BCL6, which directs the germinal-centre B cell and follicular T-helper cell programs. Conditional CUL3 deletion in mice demonstrated an essential role for CUL3 in the development of PLZF- and BCL6-dependent lineages. We conclude that distinct lineage-specific BTB-ZF transcription factors recruit CUL3 to alter the ubiquitination pattern of their associated chromatin-modifying complex. We propose that this new function is essential to direct the differentiation of several T- and B-cell effector programs, and may also be involved in the oncogenic role of PLZF and BCL6 in leukaemias and lymphomas8,9.
Similar content being viewed by others

Main
To investigate the molecular mechanisms that PLZF uses to regulate the innate-like natural killer T (NKT)-cell thymocyte differentiation program during development, we examined its protein-interaction partners. NKT-cell thymocytes were purified from Vα14-Jα18-transgenic mice and, after immunoprecipitation with an anti-PLZF antibody, associated proteins were submitted to mass spectrometry analysis (Table 1, column 1, and Supplementary Fig. 1). A major group was composed of nuclear proteins involved in binding and modifying chromatin, including HDAC1 and DNMT1, which were previously reported to interact with PLZF in myeloid cells9,10, as well as special AT-rich binding protein 1 (SATB1) and lamin B1 (LMNB1), which anchor specific DNA sequences to nuclear compartments associated with gene activation and repression, respectively11,12,13,14. We focused on the E3 ubiquitin ligase CUL3 because previous reports had established that the BTB domain of several proteins, including the BTB-ZF protein BAZF, could serve as ‘adaptors’ for CUL3-mediated ubiquitination by binding both CUL3 and its substrates3,4,5,6,7,15. Reciprocal immunoprecipitation of CUL3-associated proteins pulled down PLZF as a major protein along with an overlapping set of proteins (Table 1, column 2, and Supplementary Fig. 1). Furthermore, confocal microscopic analysis of NKT-cell thymocytes demonstrated colocalization of the two proteins in a speckled nuclear pattern (Fig. 1a, top).

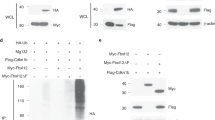
a, Confocal microscopic analysis of fresh NKT-cell thymocytes and splenic CD4 T cells from wild-type (WT) and PLZF-transgenic (tg) mice, as indicated. White colour indicates colocalization. DAPI, 4′,6-diamidino-2-phenylindole. Scale bars, 2 μm. b, Western blot analysis of anti-CUL3 and anti-PLZF immunoprecipitates (IP) from PLZF-transgenic thymocytes. 20% IN refers to 20% of the input lysate before immunoprecipitation. Data are representative of at least three independent experiments.
By contrast, in the major lineage of CD4 T cells, CUL3 was mainly found in the cytosol with only a faint presence in nuclear speckles (Fig. 1a, middle). However, after expression of a CD4 promoter-driven PLZF transgene, which induces developmental acquisition of the NKT-cell lineage effector program1,16, CUL3 was mostly in the nucleus, colocalizing with PLZF in nuclear speckles (Fig. 1a, bottom). A similar binding and transport of CUL3 from the cytoplasm to the nucleus was previously demonstrated after cotransfection with the nuclear BTB-domain-containing protein speckle-type POZ protein (SPOP) in HeLa cells17. Mass spectrometric analysis of anti-PLZF and anti-CUL3 immunoprecipitates from PLZF-transgenic thymocytes identified a similar set of proteins as that from NKT-cell thymocytes (Table 1, columns 3 and 4, and Supplementary Fig. 2), including other known partners of PLZF such as NCOR and SIN3A (ref. 9). Western blot analyses confirmed that a fraction of PLZF coprecipitated with CUL3, and that chromatin-binding and -modifying proteins such as HDAC1, SATB1 and lamin B1 were associated with the PLZF–CUL3 complex (Fig. 1b). The specificity of the interaction between PLZF and CUL3 was further tested using in vitro translated proteins, and shown to depend on the CUL3 residues Leu 52 and Glu 55 (Supplementary Fig. 3), as reported for other BTB proteins3,18, although direct binding remains to be formally established19.
Of note, the BTB-ZF transcription factor BCL6, which characterizes the germinal-centre B cell8 and follicular T-helper cell responses20 but is also transiently expressed by cortical thymocytes21, was immunoprecipitated by anti-CUL3 in thymocytes (Table 1, column 4). Analysis by western blot in transfected HeLa cells confirmed this association (Supplementary Fig. 4).
Rapid changes in ubiquitination pattern have recently been reported in chromatin-remodelling situations and are thought to regulate gene expression22,23,24. By bringing CUL3 from the cytosol to chromatin-modifying complexes in the nucleus, PLZF might be expected to induce changes in ubiquitination. This was tested using an unbiased ubiquitination proteomics method comparing whole cell lysates of thymocytes from PLZF-transgenic and wild-type littermates. Independent experiments with different batches of mice identified 48 proteins showing concordant changes, most of which consisted of increased ubiquitination in PLZF-transgenic cells (Fig. 2a and Supplementary Fig. 5). Notably, 14 of these 48 proteins were either components of the CUL3–PLZF complex identified in our previous immunoprecipitation experiments, or were well-known interaction partners of one or several of the proteins identified in the complex. The former included CAND1, LMNB1, DNMT1, SATB1 and H1.2. The latter included UHRF1, which regulates DNMT1 through ubiquitylation25; H2A.1, which is regulated by CUL3 in SPOP–CUL3 complexes26; H2A.Z, which is loaded by EP400 onto chromatin27; the deubiquitinase BRCC3 interacting with H2A, H2B, H2A.X and CAND1 (ref. 28); and the repressor ASXL1 interacting with CBX5 and EZH2 (refs 27, 29). These results are summarized in a protein-interaction diagram in Fig. 2a.

a, Mass spectrometric analysis of proteins immunoprecipitated by anti-ubiquitin branch K-GG antibodies (UbiScan) from wild-type and PLZF-transgenic thymocytes showed differential ubiquitination of the proteins depicted in blue-filled ovals in a global diagram of PLZF- and CUL3-interaction partners, with enrichment for proteins involved in chromatin organization (P < 10−6). Peptides from two independent experiments are listed in Supplementary Fig. 5. Red lines link proteins identified by coprecipitation in this study; grey lines represent previously established protein interactions (Ingenuity Pathway). b, Western blot analysis of anti-haemagglutinin (HA)–ubiquitin (Ub) immunoprecipitates from lysates of 293T cells transfected with indicated plasmids and immunoblotted with anti-SATB1 or anti-lamin B1 (LMNB1). CUL3ΔC is a mutant CUL3 lacking the E2-binding domain. The molecular mass of the lamin B1–GFP fusion protein is 75 kDa. Data are representative of at least three experiments.
Increased ubiquitination of the key nuclear proteins SATB1 and lamin B1 was directly confirmed by immunoprecipitation and western blot analyses in transfected 293T cells. In these experiments, SATB1 and lamin B1 were overexpressed because technical limitations made it impossible to determine the ubiquitination pattern of endogenous proteins (Fig. 2b). Furthermore, ubiquitination was shown to require the E2-binding domain of CUL3, supporting the conclusion that CUL3 can directly ubiquitinate these PLZF-associated proteins in vivo. In confocal microscopy experiments, PLZF and CUL3 were colocalized at lamin B1-positive sites in the nuclear lamina and at SATB1 sites of the nuclear matrix in a typical cage-like pattern (Supplementary Fig. 6). Furthermore, CUL3 was present at PLZF-bound promoters as shown by chromatin immunoprecipitation coupled to quantitative PCR (ChIP-qPCR) (Supplementary Fig. 7), suggesting that PLZF–CUL3 complexes associate at PLZF-binding promoters across the genome and in nuclear subcompartments involved in gene expression or repression.
To explore further the functional role of CUL3 in lymphocyte development and function, in which BTB-ZF transcription factors such as PLZF and BCL6 are of major importance, we bred Cul3fl/fl mice30 to CD4-Cre and CD19-Cre deleter strains. Mice lacking CUL3 in T cells exhibited a thymus with normal cellularity and representation of CD4 and CD8 T-cell lineages (Fig. 3a). T regulatory (Treg) cells showed a modest but statistically significant twofold increase (Fig. 3a). By contrast, the number of NKT cells was massively decreased, with a sharp developmental block occurring at the CD24loCD44loNK1.1− stage 1, similar to the block described in mice lacking PLZF1,2 (Fig. 3b and Supplementary Fig. 8). These results support the importance of the PLZF–CUL3 interaction in NKT-cell development.

a, Thymic subsets in Cul3cd4Δ/Δ and littermate controls. DP, double positive CD4+ CD8+. b, CD1d-PBS57 tetramer magnetic-activated cell sorting (MACS)-enriched NKT-cell thymocytes in Cul3cd4Δ/Δ, wild-type littermate (LM) controls and in Ja18−/− mice lacking NKT precursors. Top, absolute numbers of early CD24hi stage 0 precursors and maturing CD24lo cells recovered per thymus after MACS. Bottom, percentages of CD24lo stages 1, 2 and 3 based on sequential acquisition of CD44 and NK1.1. c, Splenic CD4 T-cell and B-cell counts at different ages. d, Immunohistochemical analysis of spleen from 8-week-old mice stained as indicated. Scale bars, 100 μm. e, FACS analysis of splenic subsets, as indicated, in 8-week-old mice. Numbers indicate the mean percentage ± s.e.m. f, Lethally irradiated recipients of a 3:1 mixture of wild-type and Cul3cd4Δ/Δ T cells analysed as indicated. GCB denotes B220hiIgDloGL7+Fas+; Tfh denotes CD4+PD1+CXCR5+. Data are representative of six independent experiments; n = 18–20 (e, f).
Intriguingly, in older mice lacking CUL3 in T cells, the spleen and lymph nodes became enlarged, the result of a net increase in B-cell numbers with spontaneous formation of germinal centres made of peanut agglutinin (PNA)+ B cells (Fig. 3c, d). Whereas thymic CD4+CD8− T cells exhibited a normal phenotype, a population of splenic CD4 T cells expressing a PD1+CXCR5+ follicular T-helper cell phenotype progressively accumulated in ageing mice, concomitantly with GL7+Fas+ germinal-centre B cells (Fig. 3e). Consistent with these findings, immunohistological analysis demonstrated large germinal centres with penetration of the B-cell follicles by CD4 T cells (Fig. 3d). In radiation chimaeras reconstituted with a 3:1 mixture of wild-type (CD45.1) and Cul3cd4Δ/Δ (CD45.2) T cells, the CUL3-deficient compartment showed absence of NKT cells and increased follicular CD4 T-helper cells, demonstrating the cell-intrinsic nature of these defects (Fig. 3f). As expected, germinal-centre B cells of both compartments were indiscriminately expanded. Other effector programs available to CD4 T cells, however, appeared unperturbed as CUL3-deficient CD4 cells normally expanded and differentiated towards T-helper 1 (TH1), TH2 or TH17 effector cells in vitro (Supplementary Fig. 8).
Mice lacking CUL3 in B cells showed normal development of follicular B cells but exhibited a selective four- to fivefold reduction of marginal-zone B cells in the spleen and of B1 B cells in the peritoneum (Fig. 4a). These cell-intrinsic defects were considerably amplified in the competitive environment of mixed bone marrow chimaeras (Fig. 4b, c). Whereas the circulating levels of immunoglobulins were normal or modestly decreased (Fig. 4d) and the antibody response to the T cell-independent antigen dinitrophenylated Ficoll (DNP-Ficoll) appeared conserved (Fig. 4e), T-cell-dependent B-cell responses exhibited various defects. We noted that the germinal-centre B cells that spontaneously develop in the mesenteric lymph nodes of unimmunized mice were reduced, particularly in competitive bone marrow chimaeras (Fig. 4f). The germinal-centre responses observed after immunization against the T-cell-dependent antigens nitrophenyl 23-coupled chicken γ-globulin (CGG-NP23) and sheep red blood cell (SRBC) were markedly impaired, as assessed by immunohistochemical staining of PNA+ B cells and fluorescence-activated cell sorting (FACS) staining of GL7+Fas+ B cells (Fig. 4g, h). The antibody response to SRBCs was depressed, whereas the serum antibody response to highly polyvalent nitrophenyl appeared conserved (Fig. 4g, h). These defects are similar to those reported in BCL6-deficient mice8.

a, Splenic follicular B (FOB) cells, marginal-zone B (MZB) cells and peritoneal B1 B cells in 8-week-old Cul3cd19Δ/Δ and littermate controls. b, c, FACS analysis of splenic follicular B and marginal-zone B cells (b) and peritoneal B1a/B1b subsets (c), gated as indicated, in the wild-type (CD45.1) and cd19Δ/Δ (CD45.2) compartments of 1:1 mixed bone marrow chimaeras (data representative of n = 9). d, Serum immunoglobulin isotypes in 10–11-week-old mice (n = 5). e, Serum antibody response to DNP-Ficoll at day 14. f, FACS analysis of spontaneous GCB cells in mesenteric lymph nodes (MLN) of 1:1 mixed bone marrow chimaeras (representative of n = 9). g, Splenic GCB cells (day 28) after immunization at days 0 and 21 with CGG-NP23. Top panels show FACS staining and summary bar graph (mean and s.e.m., n = 8). Bottom panels show immunohistochemical staining of spleen at day 28 and serum anti-NP25 titres at days 14 and 28. Scale bars, 100 μM. h, Similar analysis 7 days after immunization with SRBCs. Summary bar graph (mean and s.e.m., n = 15). Scale bars, 100 μM.
Our study suggests that CUL3 is an essential partner of key BTB-ZF transcription factors in the lymphoid lineage. The different effect of CUL3 on the follicular T-helper cell and the germinal-centre B-cell responses suggests that CUL3 regulates distinct components of these two BCL6-driven programs20. In addition, the defect in marginal zone B and B1 cells may hint at the existence of as yet unidentified BTB-ZF factors controlling these enigmatic populations.
Although proteomic analysis of ubiquitination was performed at the whole cell level, a considerable proportion of changes induced by PLZF expression was concentrated on the PLZF–CUL3-associated complex itself, including key nuclear matrix proteins such as SATB1 and lamin B1, which target specific DNA sequences for chromatin remodelling and gene regulation, respectively11,12,13,14. The precise nature and role of these changes remain to be determined, but the emerging evidence of the importance of ubiquitination in chromatin regulation22,23,24 suggests that they specify key aspects of the transcriptional programs directed by these transcription factors. A similar function of CUL3 may regulate the oncogenic properties of PLZF and BCL6 in leukaemias and lymphomas8,9.
Methods Summary
Mass spectrometric analysis of PLZF- and CUL3-associated proteins
NKT cells (15 × 106) purified from the thymus of Vα14-Jα18-transgenic mice, or whole thymocytes (50 × 106–75 × 106) from PLZF-transgenic mice were subjected to lysis and immunoprecipitation with anti-PLZF or anti-CUL3 antibodies, trypsin-digested and analysed by liquid chromatography–electrospray tandem mass spectrometry on a thermo LTQ Orbitrap Hybrid FT mass spectrometer.
Western blot ubiquitination assay
293T cells were lipofectamine-transfected with the plasmids pHA-Ub (2 μg), pSATB1-Myc-His (2 μg), pLMNB1-GFP (2 μg), pPLZF-Flag (0.5 μg), pCUL3-Myc (2 μg) or pCUL3ΔC (2 μg) as indicated. After 24 h, 20 μM proteasome inhibitor MG132 was added and cells were incubated for another 4–6 h. Ubiquitin conjugates were immunoprecipitated with anti-haemagglutinin agarose beads and analysed by immunoblotting with anti-SATB1 and anti-lamin B1.
Mass spectrometric analysis of ubiquitinated proteins
Total thymocytes (3 × 108) were submitted to lysis, trypsin digestion and immunoprecipitation with the anti-ubiquitin branch antibody (UbiScan, Cell Signaling Technology). Eluted peptides were characterized by tandem mass spectrometry collected with an LTQ-Orbitrap Velos Hybrid mass spectrometer (Thermo). Changes in ubiquitinated peptide levels were measured between PLZF-transgenic and wild-type thymocytes. Fold-changes above 1.45 were considered if confirmed in two independent immunoprecipitation experiments using different batches of mice.
Confocal microscopy
Purified thymocytes or lymphocytes were attached to slides, fixed with 4% paraformaldehyde in PBS and permeabilized with 0.5% Triton in PBS before staining with specific antibodies and analysis on Leica SPII-STED-CW super resolution laser scanning confocal or Olympus 1X81 laser scanning microscope using Image J software.
Mice
B6 mice bearing a targeted floxed allele of CUL3 were bred to CD4-Cre and CD19-Cre deleter mice. Controls used in the experiments are littermates with a Cre+Cul3WT/WT or Cre−Cul3fl/fl genotype.
Bone marrow chimaeras
Six-to-eight-week-old lethally irradiated (10 Gy) B6 mice were reconstituted with 2 × 106–5 × 106 bone marrow cells and examined 8–10 weeks after irradiation.
Online Methods
Mice
C57BL/6, B6.SJL-Ptprca Pep3b/BoyJ (CD45.1), Cd19-Cre (B6.129P2(C)-Cd19tm1(cre)Cgn) and Cd4-Cre (B6 Tg(cd4-cre)1Cwi) mice were obtained from Jackson Laboratories. B6.Cul3fl/fl mice30 and B6.Jα18−/− mice were bred in our colony. Among the littermates of Cul3cd4ΔΔ and Cul3cd19ΔΔ mice, we used both Cre+Cul3WT/WT and Cre−Cul3fl/fl mice as controls. All mice were raised in a specific pathogen-free environment at the University of Chicago and experiments were performed in accordance with the guidelines of the Institutional Animal Care and Use Committee.
Cell culture
HeLa and 293T cells were maintained in DMEM (GIBCO) supplemented with 10% FBS and 1% penicillin/streptomycin. KG1a cells were maintained in IMDM media (GIBCO) supplemented with 10% FBS and 1% penicillin/streptomycin. All cell lines were purchased from the American Type Culture Collection (ATCC).
Plasmids and antibodies
Plasmids used in this study are gifts from investigators or were generated in the laboratory: human pcDNA3-Myc-CUL3 (Y. Xiong); pcDNA3-DN-hCUL3Flag31 (Addgene, plasmid 15820) lacking sequences 418–760 at the carboxy terminus and serving as a catalytically inactive mutant (CUL3ΔC); pcDNA3.1-SATB1-Myc-His (R. Grosschedl); pEGFP-C3-LMNB1 (Y.-H. Fu); pFlag-CMV2-PLZF (in which PLZF was PCR-amplified from C57BL/6 thymic NKT cDNA and cloned into a Sal1 site of pFlag-CMV2).
Primary antibodies were: anti-CUL3 (C-0871 from Sigma; A301-109A from Bethyl Laboratories), anti-PLZF (monoclonal 2A9 Calbiochem; polyclonal AF2944 R&D systems), anti-BCL6 (polyclonal ab19011 Abcam; C-19 and N-3 Santa Cruz Biotechnologies; monoclonal G1191E eBioscience), antiSATB1 (L745 Cell Signaling Technology; monoclonal 14 BD Bioscience), anti-lamin B1 (monoclonal 4E4 Sigma; M-20 and S-20 Santa Cruz), anti-HA (TA-150034 Origene), anti-HDAC1 (ab-7028 Abcam). Mouse, goat or rabbit IgG (mIgG2a-ab18413, gIgG-ab37373 or rIgG-ab37415-5 Abcam). Secondary antibodies were anti-rabbit IgG-HRP (GE Healthcare or eBioscience), donkey anti-goat IgG-HRP (Santa Cruz Biotechnologies).
Immunoprecipitation of PLZF- or CUL3-associated proteins
NKT cells (15 × 106) or thymocytes (50 × 106–75 × 106) were lysed on ice for 30 min in 0.5–1 ml lysis buffer (50 mM Tris-HCl, pH 8.0, 150 mM NaCl, 5 mM EDTA, pH 8.0), 0.5% (v/v) Nonidet P-40, 1 mM dithiothreitol and 1× protease inhibitor mix (Roche). Lysates were cleared by centrifugation, pre-cleared with Protein A/G sepharose beads (Invitrogen) at 4 °C for 1 h, and incubated at 4 °C for 2 h with anti-PLZF- or anti-CUL3-bound Protein A/G Sepharose (Invitrogen) beads. Beads were washed three times with lysis buffer followed by three washes with PBS plus 0.05% Triton. Bound proteins were eluted by boiling for 5 min and resolved on 10% SDS–PAGE (BioRad). For immunoblot, the gel was transferred to nitrocellulose membrane (Transblot transfer medium, 0.45 mm, Biorad) and blotted using specific antibodies.
Mass spectrometry
For the identification of coimmunoprecipitated proteins, slices of SDS–PAGE gels stained with colloidal blue (NuPAGE, Invitrogen) were destained using 100 mM ammonium bicarbonate, pH 7.5, in 50% acetonitrile. A reduction step was performed by addition of 100 μl 50 mM ammonium bicarbonate, pH 7.5, and 10 μl of 10 mM TCEP (Tris(2-carboxyethyl)phosphine-HCl) at 37 °C for 30 min. The proteins were alkylated by adding 100 μl 50 mM iodoacetamide and allowed to react in the dark at 20 °C for 30 min. Gel slices were washed in water, then in acetonitrile and dried by SpeedVac for 30 min. Trypsin digestion was carried out overnight at 37 °C using sequencing grade modified trypsin (Promega) at a 1:50 enzyme to protein ratio in 50 mM ammonium bicarbonate, pH 7.5, and 20 mM CaCl2. Peptides were extracted from the gel pieces with 5% formic acid and dried by SpeedVac. The peptide samples were analysed by a liquid chromatography–electrospray tandem mass spectrometry (LC–MS/MS) on a Thermo LTQ Orbitrap Hybrid FT mass spectrometer. Spectra were then analysed with Mascot (Matrix Science, version3, Mascot) and Sequest (ThermoFinnigan, version v.27, rev. 11) set up to search the mus_musculus database. Peptide identifications were accepted if they could be established at greater than 95% probability. Protein identifications were accepted if they could be established at greater than 90% probability and contained at least two unique identified peptides.
UbiScan analysis
Thymocytes (3 × 108) were submitted to Cell Signaling Technology for UbiScan analysis using the ubiquitin branch antibody (1990 Cell Signaling Technology) following a method modified from ref. 32. Lysates were sonicated, cleared by centrifugation, reduced and carboxamidomethylated. Total protein for each lysate was normalized before digestion. Lysates were digested with trypsin. Peptides were separated from non-peptide material by solid-phase extraction with Sep-Pak C18 classic cartridges (Waters). Lyophilized peptides were redissolved, and ubiquitinated peptides were isolated using slurries of the ubiquitin branch antibody. Peptides were eluted from antibody-resin into a total volume of 100 μl in 0.15% trifluoracetic acid. Eluted peptides were concentrated with C18 spin tips immediately before LC–MS analysis. The samples were run in duplicate to generate analytical replicates and increase the number of MS/MS identifications from each sample. Peptides were loaded directly onto a 10 cm × 75 μm PicoFrit capillary column packed with Magic C18 AQ reversed-phase resin. The column was developed with a 72-min linear gradient of acetonitrile in 0.125% formic acid delivered at 280 nl min−1. Tandem mass spectra were collected with an LTQ-Orbitrap Velos hybrid mass spectrometer (Thermo), a top-20 method, a dynamic exclusion repeat count of 1 and a repeat duration of 30 s. Mass spectrometry spectra were collected in the Orbitrap component of the mass spectrometer, and MS/MS spectra were collected in the LTQ. MS/MS spectra were evaluated using SEQUEST 3G and the SORCERER 2 platform from Sage-N Research (v4.0, Milpitas CA). Peptide assignments were obtained using a 5% false positive discovery rate. Searches were performed against the mouse NCBI database updated on 9 June 2010. Cysteine carboxamidomethylation was specified as a static modification, oxidation of methionine residues was allowed, and ubiquitination was allowed on lysine residues. Each MS/MS spectrum arises from a parent ion observed during a survey mass spectrometry scan and can be linked to the intensity of that parent ion at its chromatographic apex, essentially measuring the abundance of the peptide in the sample. Parent ion intensities were extracted from the ion chromatogram file of each sample using proprietary software and are reported in the quantification tables. Changes in ubiquitinated peptide levels were measured by taking the ratio of raw intensities. Raw intensity values were used to calculate average values and raw ratios between samples. The raw ratios were normalized based on the median ratio found, and normalized ratios and fold-changes are reported.
Flow cytometry
CD1d-PBS57 tetramers were obtained from the NIH tetramer facility. Fluorochrome-labelled monoclonal antibodies (clones indicated in bracket) against CD4 (GK1.5), CD8a (53-6.7), TCRβ (H57-597), CD24 (M1/69), CD25 (PC61), FOXP3 (FJK-16), CD44 (IM7), NK1.1 (PK136), B220 (RA3-6B2), CxCR5, PD1 (29F.1A12), ICOS (C398.4A), IgD (11.26c.2a), Fas (JO2), GL7, CD3e, CD1d (1B1), CD21/35 (7G6), CD23(B3B4), CD45.1 (A20), CD45.2 (104), CD5 (53-7.3), CD43 (S7), CD93 (AA4.1), CD19 (ID3), IgM (11/41), CD69 (H1.2F3) and γδTCR (GL3) were purchased from e-Bioscience, BD Biosciences or Biolegend. For FOXP3 intracellular flow cytometry, cells were fixed using the permeabilization and fixation buffer (Foxp3 staining buffer set) from eBioscience. Samples were analysed on an LSRII (Becton Dickinson), or sorted on a FACS Aria (Becton Dickinson) or MoFlo (Dako Cytomation). Data were analysed using FlowJo (Tree Star).
Confocal microscopy
Purified NKT-cell thymocytes or CD4 splenocytes were attached to slides (Superfrost plus microscope slides, Fisherbrand) by cytospin, and fixed for 15 min with 4% paraformaldehyde in PBS followed by three PBS washes. Cells were permeabilized with 0.5% Triton in PBS for 10 min, washed and blocked with 10% donkey serum and 1% BSA for 1 h at room temperature before staining with anti-PLZF or anti-CUL3 for 2 h at room temperature in a humidifying chamber. After PBS washes, cells were stained with donkey anti-rabbit Alexa 488 (Invitrogen), donkey anti-goat Alexa 555 antibodies (Invitrogen) or donkey anti-mouse 647 (Invitrogen) for 30 min at room temperature. Cells were washed with PBS 0.005% Triton, then PBS, and mounted with prolong gold mounting solution (Invitrogen). Control staining included rabbit IgG and goat IgG followed by corresponding secondary antibody or secondary antibody alone. Images were captured on Leica SPII-STED-CW super resolution laser scanning confocal (×100/1.4 oil) and Olympus 1X81 laser scanning microscopes and were analysed with Image J software.
Immunoprecipitation and western blot detection of ubiquitinated proteins
293T cells grown in 6-well plates were lipofectamine-transfected with plasmids pHA-Ub (2 μg), pSATB1-Myc-His (2 μg), pLMNB1-GFP (2 μg), pPLZF-Flag (0.5 μg), pCUL3-Myc (2 μg) or pCUL3ΔC (2 μg) as indicated. The plasmid concentration was kept constant by adding pmaxGFP (Amaxa). After 24 h, 20 μM MG132 was added and cells were incubated for another 4–6 h. Cells were collected with gentle scraping and resuspended in 300 μl RIPA buffer (25 mM Tris-HCl, pH 7.6, 150 mM NaCl, 1% NP-40, 1% sodium deoxycholate, 0.1% SDS and 1× protease inhibitor). Cells were lysed by sonication (Bioruptor, Diagenode) for 12 min, with 30 s on and 30 s off. Lysates were centrifuged at 13,800g at 4 °C for 10 min to remove cell debris. One-hundred microlitres of the lysate was diluted 1:4 with RIPA buffer supplemented with 1× protease inhibitors, and incubated with 25 μl anti-HA–agarose (A2095 clone HA-7, Sigma) for 4 h. The beads were washed four times with RIPA buffer and boiled for 5 min in 50 μl SDS gel-loading buffer containing 50 mM β-mercaptoethanol at 95 °C. Samples were separated by SDS–PAGE and immunoblotted with antibodies against SATB1 or lamin B1.
Immunohistochemistry
For immunofluorescence studies, frozen OCT (Tissue-Tek)-embedded 5-μm sections of spleens were dried overnight and stained with biotinylated rat anti-B220 (RA3-6B2 BD Biosciences) and Alexa-Fluor 488-conjugated rat anti-CD4 (RM4-5 Invitrogen) antibodies, followed by Cy3-streptavidin (Invitrogen) and visualized using a SP5 II microscope (Leica). Data were analysed using ImageJ (Bitplane) software.
For immunohistochemical studies, frozen OCT-embedded sections were dried overnight, fixed with ice-cold acetone and incubated with methanol and 0.3% hydrogen peroxide to neutralize endogenous peroxidase activity, and blocked with 5% rat serum, then streptavidin-biotin blocking (Vector Laboratories). Sections were stained with biotinylated PNA (Vector Laboratories), followed by Vectastain ABC-alkaline phosphatase kit (Vector Laboratories) and the Vector Red alkaline phosphatase substrate kit (Vector Laboratories). After treatment with serum and streptavidin-biotin block, sections were stained with biotinylated rat anti-B220 antibody (RA3-6B2 BD Biosciences), followed by reaction with the Vectastain ABC kit (Vector Laboratories) and ImmPACT SG peroxidase substrate kit (Vector Laboratories) according to manufacturer’s instructions. Sections were dehydrated, cleared with xylene and mounted using Permount solution (Fischer Scientific). Micrographs were taken with the FSX-100 microscope camera system (Olympus) and data were analysed using ImageJ (Bitplane) software.
Generation of bone marrow chimaeras
Six-to-eight-week-old B6 (CD45.1) mice were subjected to irradiation with 10 Gy using a γ-cell 40 irradiator with a caesium source and were injected intravenously 3–6 h later with 2 × 106–5 × 106 bone marrow cells obtained from the femurs of donor mice. Bone marrow-reconstituted mice were analysed 8–10 weeks after irradiation
NKT cell enrichment
NKT cells were labelled with allophycocyanin (APC)-conjugated CD1d-PBS57 tetramers, bound to anti-APC magnetic beads, and enriched on an MACS cell separator (Miltenyi Biotech) as described previously33.
Mouse immunizations
Six-to-eight-week-old mice were immunized intraperitoneally with 100 μg of DNP-Ficoll (Biosearch Technologies) in PBS, 50 μg CGG-NP23 (Biosearch Technologies) mixed 1:1 with alum, or 2 × 108 SRBCs (Lampire Biological Laboratories). Mice injected with CGG-NP23 were boosted on day 21 with the same inoculum and killed on day 28. Levels of anti-DNP and anti-NP antibodies were determined by ELISA against BSA-DNP7 and BSA-NP25, respectively (Biosearch Technologies) and anti-SRBC antibodies were measured by FACS using an indirect isotype-specific immunofluorescence assay (eBiosciences, Southern Biotech).
Statistical analysis
Unpaired Student’s t-test was performed with Prism (Graph Pad Software). *P < 0.05; **P < 0.001; ***P < 0.0001.
References
Savage, A. K. et al. The transcription factor PLZF directs the effector program of the NKT cell lineage. Immunity 29, 391–403 (2008)
Kovalovsky, D. et al. The BTB-zinc finger transcriptional regulator PLZF controls the development of invariant natural killer T cell effector functions. Nature Immunol. 9, 1055–1064 (2008)
Xu, L. et al. BTB proteins are substrate-specific adaptors in an SCF-like modular ubiquitin ligase containing CUL-3. Nature 425, 316–321 (2003)
Furukawa, M., He, Y. J., Borchers, C. & Xiong, Y. Targeting of protein ubiquitination by BTB–Cullin 3–Roc1 ubiquitin ligases. Nature Cell Biol. 5, 1001–1007 (2003)
Geyer, R., Wee, S., Anderson, S., Yates, J. & Wolf, D. A. BTB/POZ domain proteins are putative substrate adaptors for cullin 3 ubiquitin ligases. Mol. Cell 12, 783–790 (2003)
Pintard, L., Willems, A. & Peter, M. Cullin-based ubiquitin ligases: Cul3-BTB complexes join the family. EMBO J. 23, 1681–1687 (2004)
Zimmerman, E. S., Schulman, B. A. & Zheng, N. Structural assembly of cullin-RING ubiquitin ligase complexes. Curr. Opin. Struct. Biol. 20, 714–721 (2010)
Basso, K. & Dalla-Favera, R. BCL6: master regulator of the germinal center reaction and key oncogene in B cell lymphomagenesis. Adv. Immunol. 105, 193–210 (2010)
McConnell, M. J. & Licht, J. D. The PLZF gene of t(11;17)-associated APL. Curr. Top. Microbiol. Immunol. 313, 31–48 (2007)
Guidez, F. et al. RARα-PLZF overcomes PLZF-mediated repression of CRABPI, contributing to retinoid resistance in t(11;17) acute promyelocytic leukemia. Proc. Natl Acad. Sci. USA 104, 18694–18699 (2007)
Yasui, D., Miyano, M., Cai, S., Varga-Weisz, P. & Kohwi-Shigematsu, T. SATB1 targets chromatin remodelling to regulate genes over long distances. Nature 419, 641–645 (2002)
Cai, S., Lee, C. C. & Kohwi-Shigematsu, T. SATB1 packages densely looped, transcriptionally active chromatin for coordinated expression of cytokine genes. Nature Genet. 38, 1278–1288 (2006)
Reddy, K. L., Zullo, J. M., Bertolino, E. & Singh, H. Transcriptional repression mediated by repositioning of genes to the nuclear lamina. Nature 452, 243–247 (2008)
Zullo, J. M. et al. DNA sequence-dependent compartmentalization and silencing of chromatin at the nuclear lamina. Cell 149, 1474–1487 (2012)
Ohnuki, H. et al. BAZF, a novel component of cullin3-based E3 ligase complex, mediates VEGFR and Notch cross-signaling in angiogenesis. Blood 119, 2688–2698 (2012)
Savage, A. K., Constantinides, M. G. & Bendelac, A. Promyelocytic leukemia zinc finger turns on the effector T cell program without requirement for agonist TCR signaling. J. Immunol. 186, 5801–5806 (2011)
Kwon, J. E. et al. BTB domain-containing speckle-type POZ protein (SPOP) serves as an adaptor of Daxx for ubiquitination by Cul3-based ubiquitin ligase. J. Biol. Chem. 281, 12664–12672 (2006)
Wimuttisuk, W. & Singer, J. D. The Cullin3 ubiquitin ligase functions as a Nedd8-bound heterodimer. Mol. Biol. Cell 18, 899–909 (2007)
Errington, W. J. et al. Adaptor protein self-assembly drives the control of a Cullin-RING ubiquitin ligase. Structure 20, 1141–1153 (2012)
Crotty, S. Follicular helper CD4 T cells (TFH). Annu. Rev. Immunol. 29, 621–663 (2011)
Hyjek, E., Chadburn, A., Liu, Y. F., Cesarman, E. & Knowles, D. M. BCL-6 protein is expressed in precursor T-cell lymphoblastic lymphoma and in prenatal and postnatal thymus. Blood 97, 270–276 (2001)
Braun, S. et al. The Cul4-Ddb1Cdt2 ubiquitin ligase inhibits invasion of a boundary-associated antisilencing factor into heterochromatin. Cell 144, 41–54 (2011)
Wang, H. et al. Role of histone H2A ubiquitination in Polycomb silencing. Nature 431, 873–878 (2004)
Bosch-Presegué, L. et al. Stabilization of Suv39H1 by SirT1 is part of oxidative stress response and ensures genome protection. Mol. Cell 42, 210–223 (2011)
Du, Z. et al. DNMT1 stability is regulated by proteins coordinating deubiquitination and acetylation-driven ubiquitination. Sci. Signal. 3, ra80 (2010)
Hernández-Muñoz, I. et al. Stable X chromosome inactivation involves the PRC1 Polycomb complex and requires histone MACROH2A1 and the CULLIN3/SPOP ubiquitin E3 ligase. Proc. Natl Acad. Sci. USA 102, 7635–7640 (2005)
Hargreaves, D. C. & Crabtree, G. R. ATP-dependent chromatin remodeling: genetics, genomics and mechanisms. Cell Res. 21, 396–420 (2011)
Sowa, M. E., Bennett, E. J., Gygi, S. P. & Harper, J. W. Defining the human deubiquitinating enzyme interaction landscape. Cell 138, 389–403 (2009)
Beisel, C. & Paro, R. Silencing chromatin: comparing modes and mechanisms. Nature Rev. Genet. 12, 123–135 (2011)
McEvoy, J. D., Kossatz, U., Malek, N. & Singer, J. D. Constitutive turnover of cyclin E by Cul3 maintains quiescence. Mol. Cell. Biol. 27, 3651–3666 (2007)
Jin, J., Ang, X. L., Shirogane, T. & Wade Harper, J. Identification of substrates for F-box proteins. Methods Enzymol. 399, 287–309 (2005)
Rush, J. et al. Immunoaffinity profiling of tyrosine phosphorylation in cancer cells. Nature Biotechnol. 23, 94–101 (2005)
Benlagha, K., Wei, D. G., Veiga, J., Teyton, L. & Bendelac, A. Characterization of the early stages in thymic NKT cell development. J. Exp. Med. 202, 485–492 (2005)
Acknowledgements
We thank A. Dinner, J. Licht, G. Prive, A. Ruthenburg, R. Sciammas, H. Singh and P. Wilson for discussions, J. C. Silva and M. Stokes for UbiScan analysis, C. Labno and V. Bindokas for help with confocal microscopy, and K. Block, L. Roach, D. Zabner, F. Meng and L. Bai for help with experiments. This work was supported by National Institute of Health (NIH) grants 5RO1GM082940 (J.D.S.) and RO1AI038339 (A.B.), and an Irvington Institute postdoctoral fellowship from the Cancer Research Institute (R.M.). A.B. is a Howard Hughes Medical Institute Investigator.
Author information
Authors and Affiliations
Contributions
R.M. designed the research, performed experiments and analysed data. M.P.S., S.T.S., A.M., M.G.C. and C.B.-V. performed experiments and analysed data. J.D.S. helped to design experiments and provided the Cul3fl/fl mice and CUL3 constructs. R.M. and A.B. co-wrote the paper. A.B. supervised the research.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing financial interests.
Supplementary information
Supplementary Information
This file contains Supplementary Figures 1-8, Supplementary Methods and a Supplementary Reference. (PDF 1858 kb)
Rights and permissions
About this article
Cite this article
Mathew, R., Seiler, M., Scanlon, S. et al. BTB-ZF factors recruit the E3 ligase cullin 3 to regulate lymphoid effector programs. Nature 491, 618–621 (2012). https://doi.org/10.1038/nature11548
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/nature11548
This article is cited by
-
The dual roles of autophagy and the GPCRs-mediating autophagy signaling pathway after cerebral ischemic stroke
Molecular Brain (2022)
-
Leukemia/lymphoma-related factor (LRF) or osteoclast zinc finger protein (OCZF) overexpression promotes osteoclast survival by increasing Bcl-xl mRNA: A novel regulatory mechanism mediated by the RNA binding protein SAM68
Laboratory Investigation (2022)
-
The CRL3BTBD9 E3 ubiquitin ligase complex targets TNFAIP1 for degradation to suppress cancer cell migration
Signal Transduction and Targeted Therapy (2020)
-
The diverse roles of SPOP in prostate cancer and kidney cancer
Nature Reviews Urology (2020)
-
Cullin-3: Renal and Vascular Mechanisms Regulating Blood Pressure
Current Hypertension Reports (2020)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
